Reduce the quantification time of 13C 1000 times by including polarization transfer and indirect detection.
The most common approach to quantifying materials by NMR utilizes 1H NMR spectra, which combines high intrinsic sensitivity with straightforward, inherently quantitative signal integrals. In complex formulations and reaction mixtures, however, 1H spectra often suffer from peak overlap, which limits the accuracy to quantitate the different components. A way to minimize overlap is to measure 13C spectra. The 13C chemical shift spreading is far wider than that of 1H, and, with 1H decoupling, 13C resonances appear as simple singlets, which greatly simplifies assignment and integration. The drawback is sensitivity: the low natural abundance of 13C (≈1.1%) and its typically long T₁ relaxation times make direct 13C acquisition time-consuming and, for many samples, impractical. Polarization transfer addresses this limitation by using polarization from abundant 1H spins to boost 13C signal-to-noise. Schemes such as NOE, INEPT/DEPT, and related variants can yield up to ~4-fold increases in apparent 13C sensitivity and allow repetition delays to be governed by the shorter 1H T₁ rather than the longer 13C T1, accelerating data collection. The common perception, however, is that polarization-transfer experiments are not quantitative because the transfer efficiencies vary across different carbon sites and molecular environments, so direct comparison of peak areas to an internal standard no longer reports concentration in a site-independent way. We have recently shown that this limitation can be overcome. Under fixed acquisition conditions, transferred 13C signal amplitudes remain proportional to analyte concentration; by calibrating each target signal against external standards containing known concentrations, one obtains linear response curves that restore quantitative accuracy without sacrificing the sensitivity benefits of polarization transfer. In this application note, we extended the approach to the more sensitive two-dimensional HSQC sequence, where polarization is transferred back to 1H for detection, leveraging 1H sensitivity with an up to 32-fold increase in SNR relative to the 13C inverse-gated experiment. This method also benefits from shorter measurement times due to the shorter 1H T1 enhancing the sensitivity of 13C detection for quantitative measurements in scenarios where conventional 1H quantitation is compromised by spectral overlap.
Table 1 summarizes the advantages and disadvantages of the three approaches.

Table 1. Advantages and disadvantages of different qNMR approaches.
Experimental Setup
Experiments have been performed on a Spinsolve 90 ULTRA Multi-X with 13C available on the X-channel. Spectra were acquired for samples containing 10, 20, 50, 100, 200, 500 and 1000 mM lidocaine in CDCl3. For a fair comparison, experimental parameters were set to yield equal acquisition times for DEPT and HSQC, i.e. an HSQC with 128 increments and 4 scans x 2 (for phase-sensitive detection) would be the equivalent of a DEPT with 1024 scans. DEPT spectra were acquired as DEPT-45. The HSQCs were acquired with an echo-antiecho sequence. Repetition times of 2, 5 and 10 seconds were assessed, with 5 s being a good compromise for experiment time and quantitative measurement condition. For the analysis, the singlet for the aromatic CH3 groups at 2.3 / 18.7 ppm was integrated, with horizontal traces extracted and integrated in the HSQC and direct integration in the DEPT.
NMR Results & Discussion
Fig. 1 shows the structure of lidocaine as well as representative DEPT and HSQC spectra as well as a vertical HSQC trace, with signals used for quantitation and respective signal-to-noise ratios (SNR) highlighted in blue. For the DEPT the limit of detection is around 23 mM, while for the HSQC it is ~3 mM.
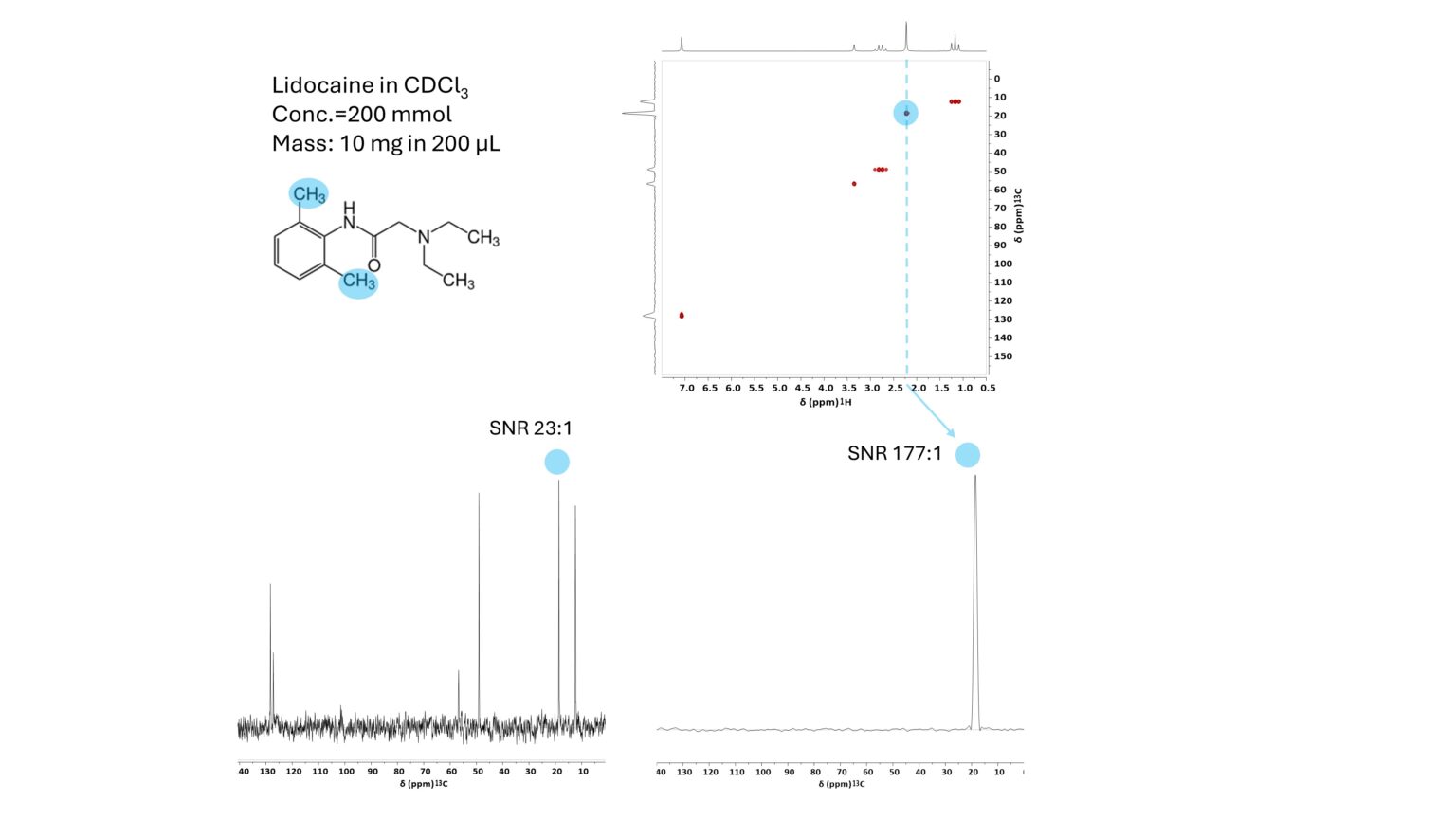
Figure 1. Structure of lidocaine (top left), HSQC (top right), 13C DEPT-45 (bottom left) and vertical traces of HSQC (bottom right) for the aromatic CH3 groups of lidocaine (200 mM in CDCl3). Signals used for quantitation and their respective SNR are highlighted in blue.
The theoretical maximum improvement in SNR over 13C{1H} is governed by the gyromagnetic ratios of 1H and 13C. For the DEPT experiment we get an improvement of 4 due to the polarization transfer from 1H to 13C:
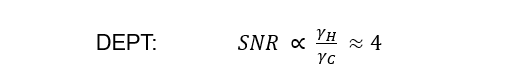
For the HSQC with 1H excitation and detection the improvement over DEPT is about eight-fold, or 32-fold over a 1D 13C{1H} inverse gated experiment:
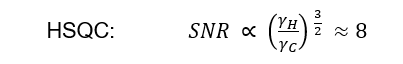
The advantage of the HSQC lies in the significant reduction in measurement time, when compared to the 13C{1H} inverse gated experiment, a 32-fold higher SNR corresponding to a 1000-fold shorter experiment time for the same concentration. The enhancement observed experimentally between DEPT and HSQC is about 7.7-fold, which is really close to the theoretical value. Figure 2 compares the concentration dependence of the DEPT and HSQC spectra for the aromatic CH3 groups for the two signals. Both DEPT and HSQC signals yield the expected linear response as a function of concentration. However, thanks to the superior SNR of the HSQC a much smaller deviation from linearity is observed.
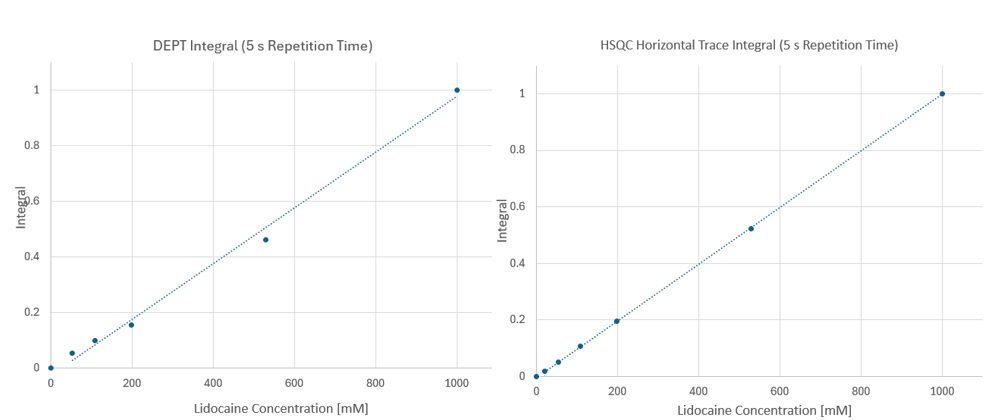
Figure 2. Concentration dependence of DEPT (left) and horizontal HSQC trace (right) integrals for the aromatic CH3 singlet of lidocaine.
Conclusion
The data show that either DEPT or HSQC can be used for quantitation. However, the HSQC response is far superior in SNR, allowing extension of the concentration range at the lower end, and should be the preferred choice if suitable signals, preferably singlets can be identified. The SNR enhancement obtained with the HSQC makes it possible to reduce the measurement time by a factor of 64 compared to the time required by the DEPT sequence and a factor about 1000 compared to the quantitative inverse gated 13C{1H} experiment.
Read the full application note – CLICK HERE