
Quantification using any analytical method is no more than calibrating an instrumental response with a known reference, and then calculating the concentration of an unknown sample from the measured instrument response. One method of calibrating the NMR spectrometer is with an internal standard. Both the sample and the reference are weighed out and co-dissolved into a single solution. The integral of the peak associated with the reference sample is used to calibrate the instrument response. One or more peaks associated with the sample of interest are then used to determine the sample concentration or purity. The purity can be calculated with the following equation.

Where:
- M =Molecular weight
- I = Integral
- N = Normalisation factor (the number of nuclei represented by the peak)
- m = Mass
- P = Purity
- Sample = designates the sample of interest
- Ref = Reference Standard
Methylsulfonyl methane
When quantitative methods are validated there are standard requirements for accuracy, precision, range, and linearity over that range. Table 1 below shows general requirements for a Category I NMR method when measuring a drug substance (there are other specifications for finished products and impurities).
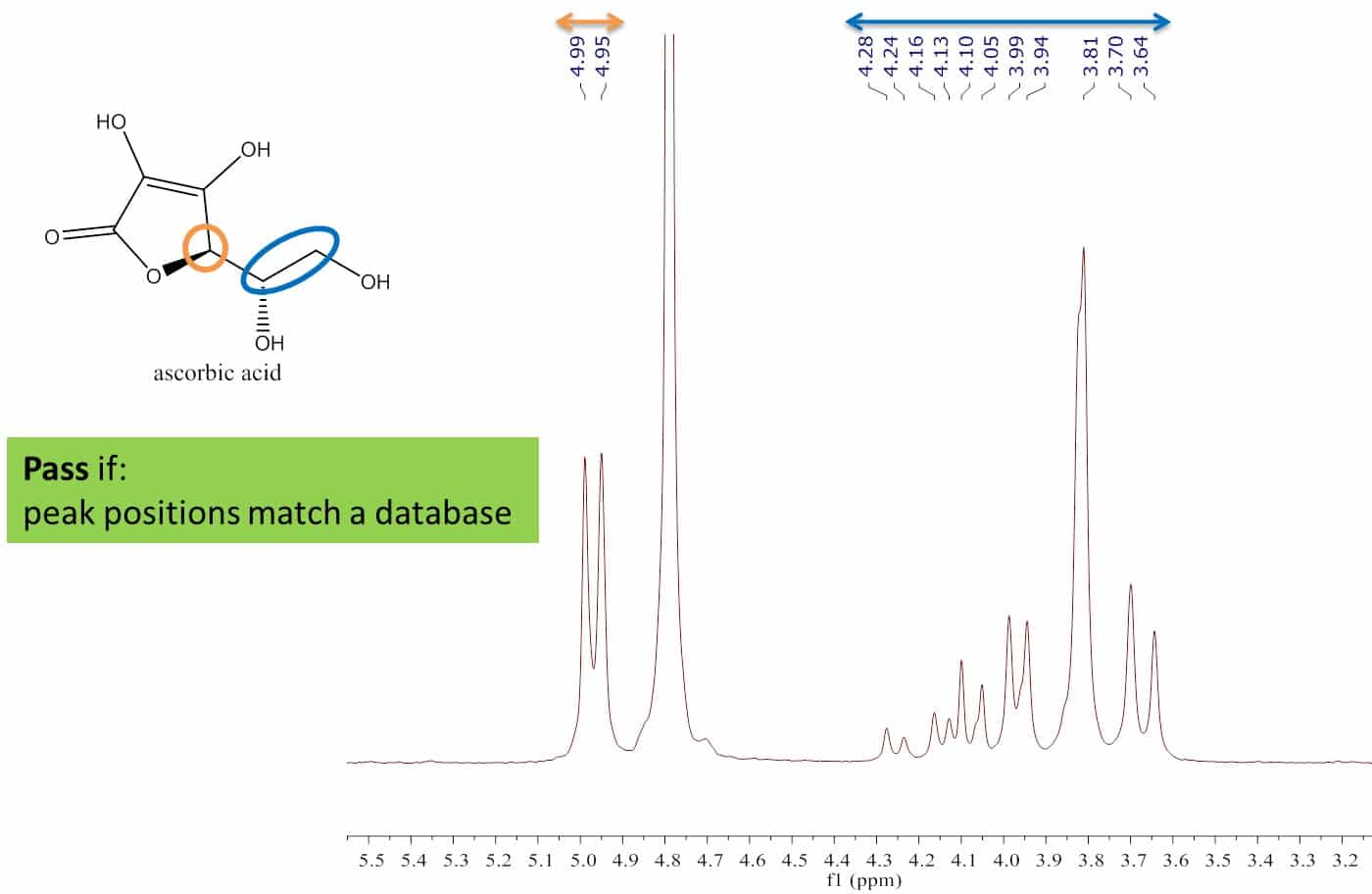
These requirements were tested by measuring the purity of one reference standard, methylsulfonylmethane (MSM), with another, maleic acid. Maleic acid is a common reference standard so this was used as the reference to measure the known purity of MSM (99.5%). A spectrum of the mixture in D2O is shown in Figure 3.




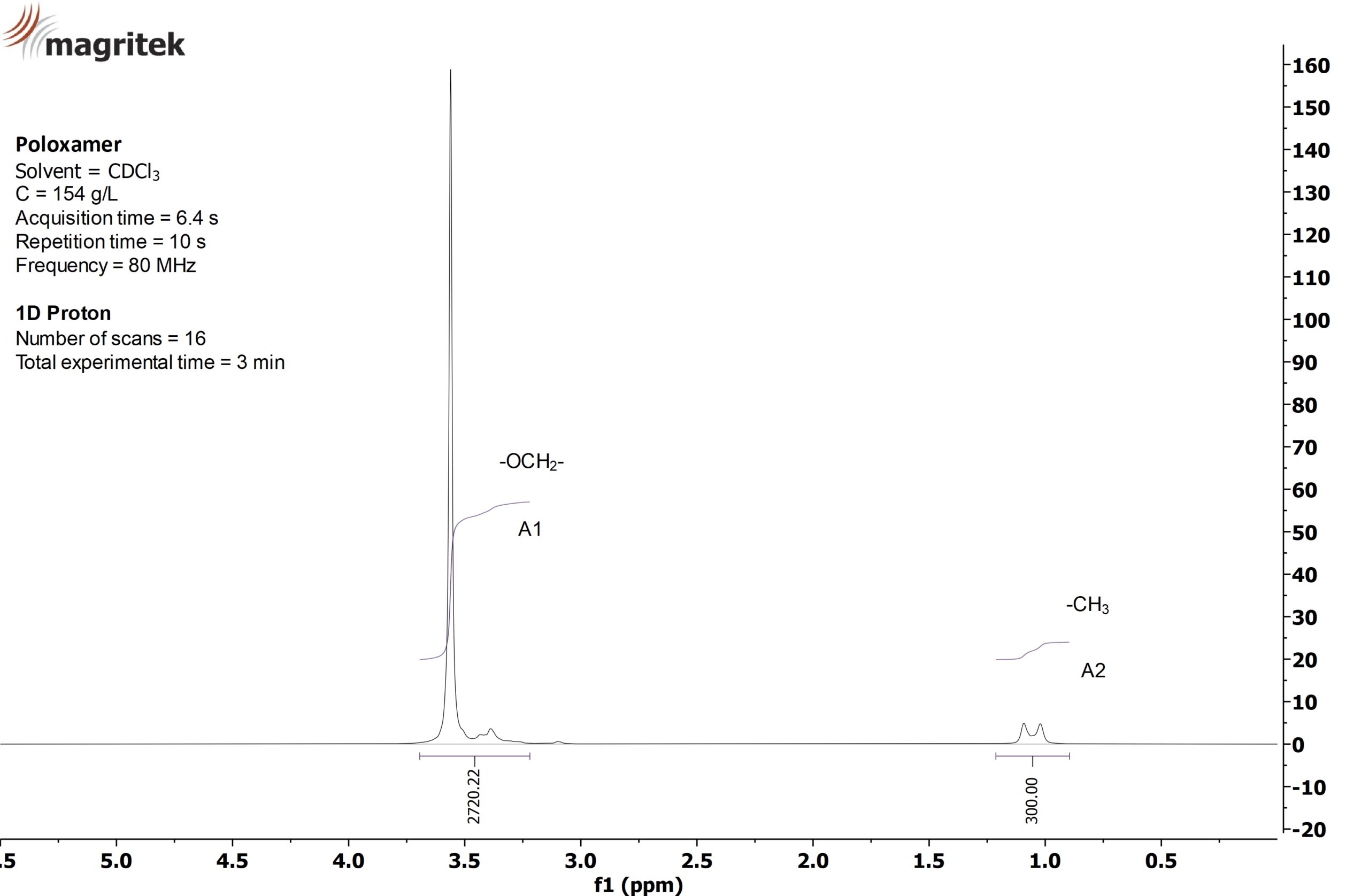
Poloxamers are oxyethylene-oxypropylene block copolymers commonly used as non-ionic surfactants. The ratio of oxyethylene to oxypropylene is a tuneable chemical property that is commonly determined by NMR spectroscopy. The peak at 1 ppm is from the propylene methyl and the peak at 3.5 ppm is from the polymer backbone. The weight percent oxyethylene is calculated using the following equations:

The “7” in F127 means that the sample has approximately 70% oxyethylene and the NMR determined it to be 74%.





