Simultaneous
Dual-Phase Reaction Monitoring by Benchtop NMR
Real-Time Kinetic Analysis of a Biphasic Suzuki–Miyaura Coupling Reaction

Introduction
In the past decade, benchtop nuclear magnetic resonance (NMR) spectroscopy has become a well-established tool for on-line reaction monitoring. The compact size and low weight of these spectrometers provide the mobility required to install them directly inside a fume hood or on a laboratory bench, in proximity to the chemical reactor. In particular, recent generations of Spinsolve ULTRA benchtop NMR spectrometers eliminate the need for sample preparation before analysis, enabling chemists to pump the reaction mixture directly from the reactor through the NMR spectrometer for real-time analysis. Spinsolve ULTRA spectrometers deliver exceptional magnetic field homogeneity, allowing for highly selective suppression of signals from protonated solvents. This method enables the acquisition of high-quality NMR spectra without requiring time-consuming sample workup to replace conventional solvents with deuterated ones. In addition, benchtop NMR allows the observation of multiple nuclei, present in reacting molecules. The new Multi Xn electronic platform enables automatic switching between different nuclei, facilitating multinuclear experiments without user intervention.
The performance of benchtop NMR for reaction monitoring has been extensively demonstrated through successful integration with both continuous-flow and batch reactor setups. Literature examples to date report reactions carried out in a single solvent[1-4]. However, in a good number of cases, reactions are performed in biphasic systems[5] composed of two immiscible solvents -typically an aqueous and an organic phase- mixed to enhance reaction efficiency and selectivity or to simplify downstream separation and purification. In such systems, starting materials, products, and side products may partition between both phases. Consequently, a complete understanding of reaction kinetics and mechanisms requires simultaneous monitoring of the concentrations of all relevant species in each phase.
For example, inorganic bases or polar reagents may be confined to the aqueous phase, while organic substrates and products preferentially reside in the organic phase. In transition-metal-catalyzed cross-coupling reactions, the catalyst may be selectively retained in one phase, facilitating separation of the product and enabling catalyst reuse. Furthermore, continuous extraction of the product into one phase can shift the reaction equilibrium, suppress side reactions, or reduce catalyst deactivation.
However, the performance of biphasic systems depends critically on mass transfer across the liquid–liquid interface and on the dynamic distribution of reactants, intermediates, catalysts, and products between the two phases. Since chemical species may partition differently over the course of the reaction, monitoring only one phase provides an incomplete and potentially misleading picture of the reaction kinetics and mechanism. A comprehensive understanding therefore, requires simultaneous quantification of all relevant species in both phases.
From an analytical perspective, on-line monitoring of biphasic systems requires reliable and continuous phase separation prior to analysis, as well as dedicated in-line analytical instrumentation for each phase. Recently, Hein and co-workers[6] reported a practical approach for phase-selective sampling using a polyethylene filter to extract the organic phase and an activated stainless-steel filter to isolate the aqueous phase, achieving efficient separation of water/organic solvent mixtures under flow conditions. In their study, however, only one of the separated phases was analyzed by high-field NMR spectroscopy. As a result, the reaction had to be repeated to obtain data for the second phase, preventing true simultaneous monitoring and limiting direct kinetic correlation between phases.
In this application note, we leverage the flexibility, compactness, and robustness of benchtop NMR spectroscopy to present the first experimental setup that enables simultaneous on-line monitoring of both phases of a biphasic reaction system within a single experiment. By integrating phase-selective extraction with two independent benchtop NMR spectrometers, both solvent layers can be continuously analyzed in real time, eliminating the need for repeated experiments and enabling direct comparison of concentration profiles across phases.
The capabilities of this setup are demonstrated using a Suzuki–Miyaura coupling reaction performed under biphasic conditions. For the first time, benchtop NMR spectroscopy is used to non-destructively and quantitatively monitor the reaction kinetics in both phases simultaneously. This approach provides detailed insight into the formation and consumption of reactants, intermediates, side products, and the desired coupling product, while also revealing the time-dependent distribution of chemical species between the aqueous and organic phases. Such information is essential for understanding interphase mass transfer, catalyst speciation, and selectivity-determining steps in biphasic catalysis.
These results are enabled by the high stability, sensitivity, and excellent magnetic field homogeneity of the Spinsolve ULTRA spectrometer, which allows accurate quantification directly in conventional (non-deuterated) solvents. Moreover, the fully in-line configuration permits operation under inert conditions, minimizing oxygen exposure associated with manual sampling and preserving sensitive catalytic systems.
Experiments and Results
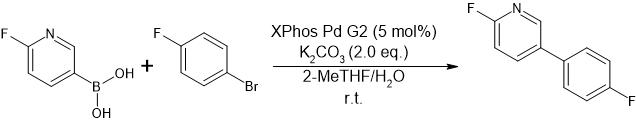
Figure 1: Suzuki-Miyaura-Coupling of 6-Fluoro-3-pyridinylboronic acid and 1-Bromo-4-fluorobenzene.
To demonstrate the performance of the biphasic reaction monitoring platform, the Suzuki–Miyaura cross-coupling reaction shown in Figure 1 was investigated. In this system, 6-fluoro-3-pyridinylboronic acid and 1-bromo-4-fluorobenzene are initially distributed in different solvent phases to establish a well-defined biphasic setup prior to catalyst addition. The reaction was performed using 2-methyltetrahydrofuran (2-MeTHF) as the organic phase and water as the aqueous phase. After combining the two immiscible solvents, 6-fluoro-3-pyridinylboronic acid was introduced into the mixture. Although this boronic acid exhibits partial solubility in the organic solvent, the subsequent addition of an aqueous potassium carbonate (K₂CO₃) solution promotes its deprotonation and transfer into the aqueous phase, where it is preferentially dissolved as the corresponding boronate species.
Following establishment of this phase distribution, 1-bromo-4-fluorobenzene was added and remained predominantly in the organic phase due to its hydrophobic character. At this stage, the two reactants were located in separate solvent layers (this experiment is shown in another application note). The reaction was initiated by addition of the XPhos–palladium catalyst to the biphasic mixture, enabling cross-coupling at the liquid–liquid interface under continuous stirring.
To monitor the reaction, we used two Spinsolve 90 MHz ULTRA Multi X systems capable of measuring Proton, Fluorine, Carbon, Boron and Phosphorus. Both were equipped with a reaction monitoring kit 2 (RMkit 2), which consists of a glass flowcell, a peristaltic pump and HPLC tubing with the respective connectors. The setup is shown in Figure 2, where the reactor containing the mixture of the two phases and the two filters can also be seen.

Figure 2: Setup used to monitor the concentration of substrates, intermediates and product reacting in a biphasic solvent mixture. The two immiscible solvents (organic and aqueous phases) are continuously stirred during the reaction but can be isolated by means of two inline filters. A polyethylene filter QLA FIL 010-PT-a is used to extract the organic phase, while a stainless-steel filter Scipro A-302A is used to isolate the aqueous phase. The filters are connected to two independent reaction monitoring kits RMK2 mounted in two Spinsolve 90 ULTRA Multi X spectrometers for simultaneous acquisition on NMR spectra.
To separate the two mixed phases, two different filters were used. To isolate the organic and the aqueous phase a polyethylene filter (QLA FIL 010-PT-a, 10 micrometer pore size, 1/8” ID UHMW) and a stainless steel (SS) sintered metal filter (Scipro: A-302A, Solvent Filter inlet 1/4-28, freshly activated) were used, respectively. The stainless-steel filter was activated by immersing it in 5 M K2CO3 solution for some minutes under sonication and washing it afterwards with deionized water and air drying. This procedure was repeated three times before it was connected to the reaction-monitoring tubing.
The reaction was carried out in a 100 mL three-necked round-bottom flask equipped with a magnetic stirrer. The reaction mixture was prepared by dissolving 2.11 g (15.0 mmol) 6-Fluoro-3-pyridinylboronic acid in 60 mL 2-MeTHF and 60 mL water. To transfer 6-Fluoro-3-pyridinylboronic acid to the aqueous phase, 6.0 mL (30.0 mmol) of a 5.0 M K2CO3 was added to the solution. Afterwards, we added 2.63 g (1.65 mL, 15.0 mmol) 1-Bromo-4-fluorobenzene. Before the addition of the Palladium catalyst, Nitrogen was bubbled for 15 min through the solvents to remove the oxygen dissolved in it and avoid the direct graduation of the palladium catalyst after its addition. Figure 3 shows an example of the solvent mixture without stirring (left), where two immiscible phases can be identified. When stirred (right) the two phases are mixed to form a cloudy temporary emulsion.

Figure 3: Separated phases (left photo) of the solvents and mixed phases under stirring (right photo) as an example.
The RMX module in the Spinsolve software was set to monitor the reaction in continuous flow mode. In both Spectrometers the monitoring loop was set to acquire four experiments in an interleaved way. The loop included a 1D fluorine with proton decoupling (HDEC) (16 scans, 3.2 s acquisition time, and 15 seconds repetition time), a 1D boron spectrum (1024 scans, 50 ms acquisition time, and 0.2 seconds repetition time), and a 1D proton with solvent suppression (4 scans, 3.2 s acquisition time, and 15 seconds repetition time). The WET suppression sequence was set to selectively excite the water peak for the aqueous phase and all three peaks of 2-MeTHF for the organic phase.
The monitoring loop was started before adding the catalyst. In this way, the first spectra served as calibration measurements to convert signal amplitudes to concentration during the reaction. Subsequently, after adding the catalyst, the concentration of the starting materials, product, and side products can be monitored in real time by detecting the signal of the different nuclei present in each structure. For this particular reaction, both starting materials and product contain fluorine, which is a very convenient nucleus to quantify the different species. Figure 4 shows a stack plot of the 19F spectra collected for the organic phase during the full reaction. The signal of 1-Bromo-4-fluorobenzene is observed in the red region at -114.5 ppm and both product signals at -69.7 ppm and -113.5 ppm are marked in violet and green. The violet one belongs to the fluorine close to the nitrogen and the green one belongs to the fluorine located at the other aromatic ring. In the blue region, we can also identify the signal of the residual 6-Fluoro-3-pyridinylboronic acid present in the organic phase at a very low concentration. A similar data set is available for the aqueous phase, where only the signal of 6-Fluoro-3-pyridinylboronic acid is detected (see Fig. 6).
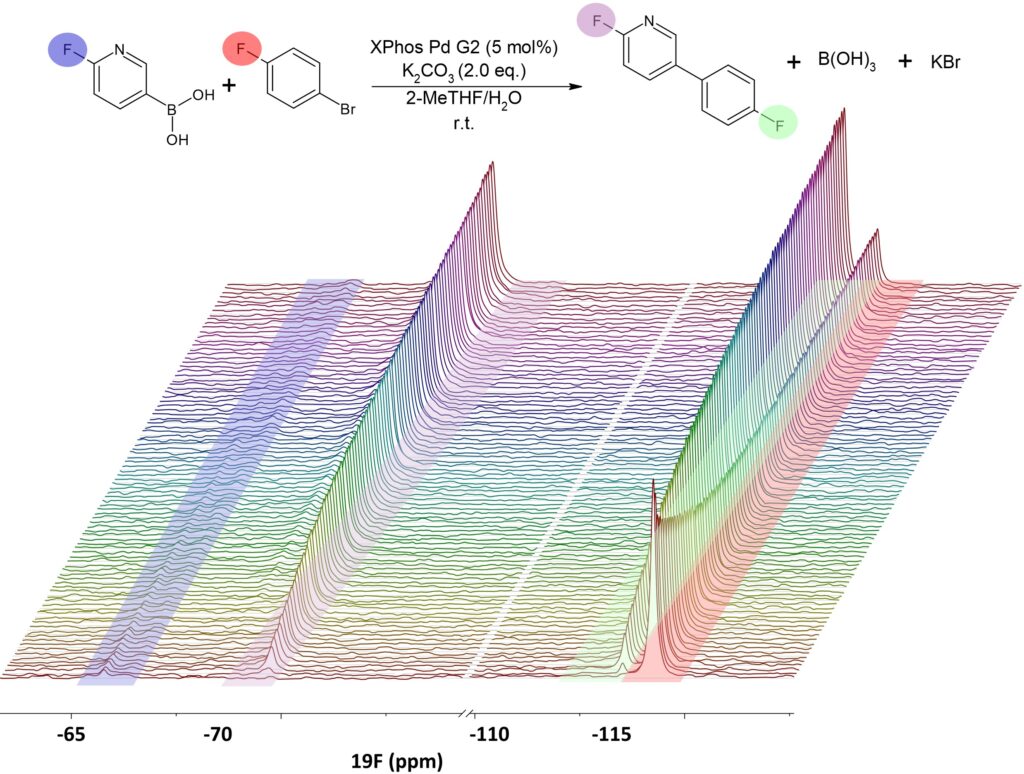
Figure 4: Stack plot showing the 19F-Spectra collected from the organic phase during the full Suzuki-Miyaura Coupling reaction The signals of 6-Fluoro-3-pyridinylboronic acid (residual) and 1-Bromo-4-fluorobenzene, as well as the two signals of the product present in the organic phase, are marked in different colors.
The concentrations calculated from the integrals of the different signals in both phases are shown in Figure 5. The two fluorine signals of the product (violet and green curves) deliver exactly the same concentration, showing how reproducible the integration is. The concentration of the starting material 1-Bromo-4-fluorobenzene starts at the prepared concentration of 250 mM and decays during the reaction to reach a plateau at about 70 mM. On the other hand, the 6-Fluoro-3-pyridinylboronic acid, which we follow in the aqueous phase, shows at the beginning a concentration of 180 mM and decay during the reaction until it is fully consumed. This difference explains why 1-Bromo-4-fluorobenzene is not consumed completely. There is also a very low concentration of the Boronic acid available in the organic phase, shown in blue in Figure 5.
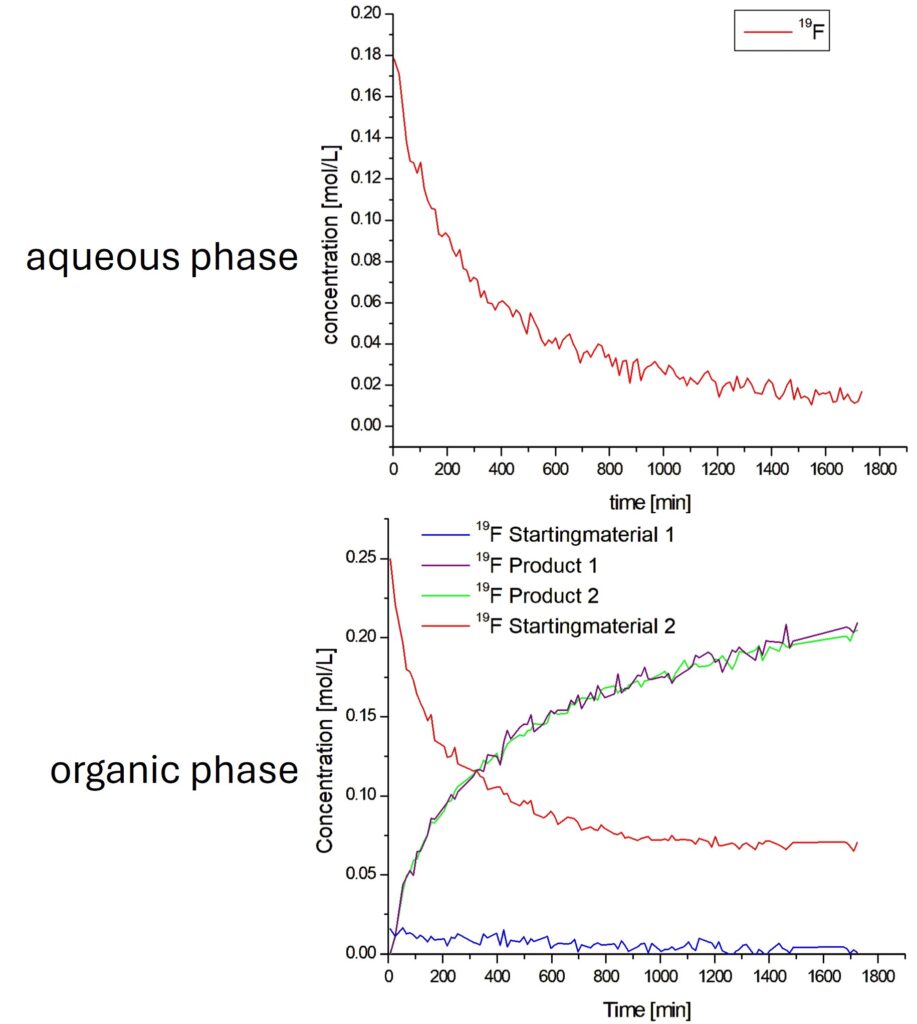
Figure 5: Calculated concentrations for each fluorine signal of the respective starting material and product during the Suzuki-Miyaura Coupling of 6-Fluoro-3-pyridinylboronic acid, detected in the aqueous phase, and 1-Bromo-4-fluorobenzene, detected in the organic phase.
The Spinsolve Multi-Xn systems are able to measure different nuclei without manually tuning and matching the probe. The spectrometer automatically retunes the probe from nucleus to nucleus by means of electronic switches controlled by the software. By measuring different nuclei, we can detect the different chemical species by choosing the most convenient nuclei for each case. In this way, we can increase chemical selectivity by choosing to follow the signal of nuclei that can be better detected and resolved. In particular, at benchtop NMR frequencies, the signals from starting materials, intermediates, and products may overlap in the proton spectra. In this situation, having alternative nuclei available facilitates the monitoring of the different species. This is the case for boronic acid. The concentration of this molecule can be monitored by detecting protons, fluorine or boron. Figure 6 shows how the consumption of boronic acid can be followed with all three nuclei present in this molecule. Moreover, boric acid formed as a byproduct of the reaction can only be detected via boron NMR (marked in green in the boron spectra). Without the boron capability this byproduct could not be detected by proton NMR. This shows the additional advantage of the MULTI-Xn systems.
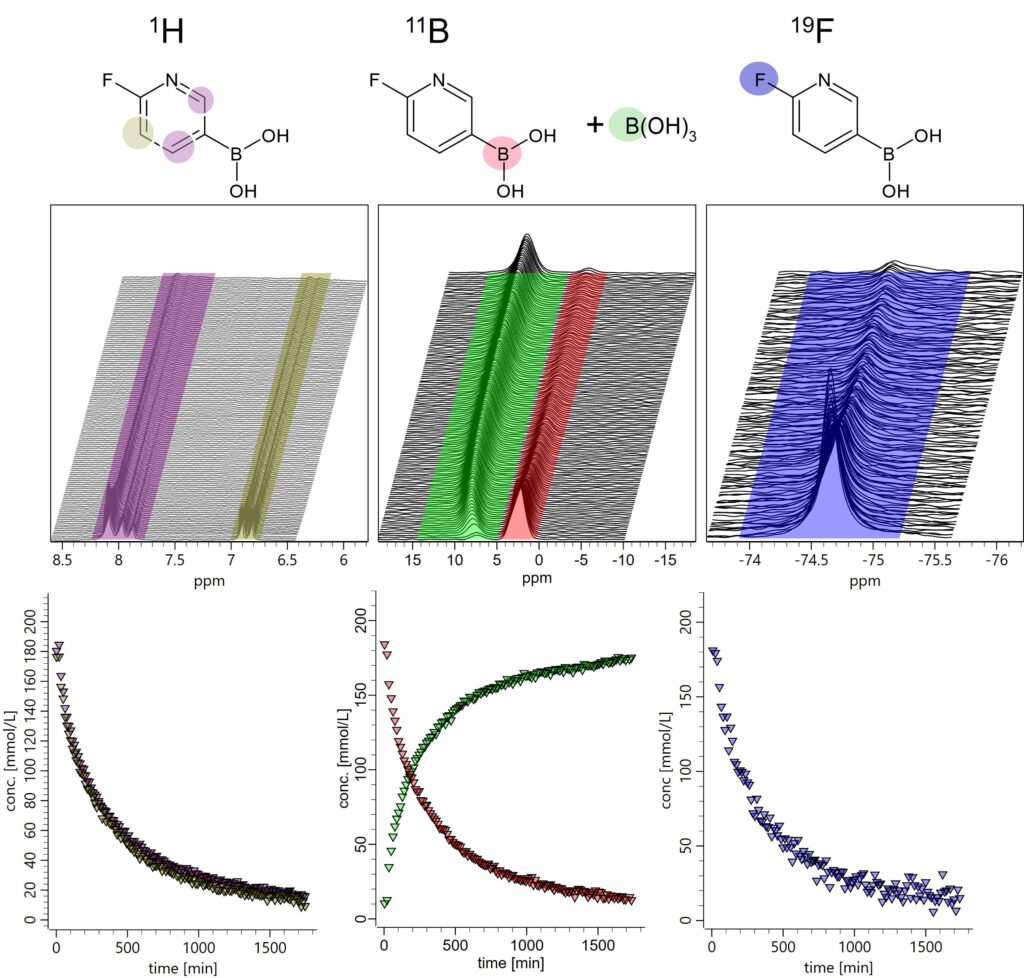
Figure 6: Stackplots of the Proton (left), Boron (center), and Fluorine (right) spectra collected for the aqueous phase during the reaction. The concentration of 6-Fluoro-3-pyridinylboronic acid dissolved in this phase decreases with time. The concentration of boric acid, on the other hand, can only be monitored from the boron spectra (green).
All nuclei measured for the aqueous phase are visible in Figure 6. Since in the aqueous phase only the boronic acid as starting material and the boric acid as a byproduct are soluble, there are not so many signals that can overlap in the proton spectrum (left plots in Fig. 6). Two different signals can be detected for the protons of the heteroaromatic ring of the boronic acid (violet and brown). Both deliver the same concentrations and can be used to follow with high accuracy the consumption of the starting material during the reaction. Boron spectra (center in Fig. 6) show both, boronic acid and boric acid, which is generated as side product, marked in red and green, respectively. The fluorine spectrum is much simpler, a single signal for boronic acid is detected at -74.5 ppm (blue region on the right plot of Fig. 6). From each stack plot, concentration curves for boronic acid were extracted and plotted together in Figure 7. The different data sets were calibrated with the signal of the known initial concentration in the mixture. The concentration curves extracted from all three nuclei closely overlap showing how linear and stable NMR is.
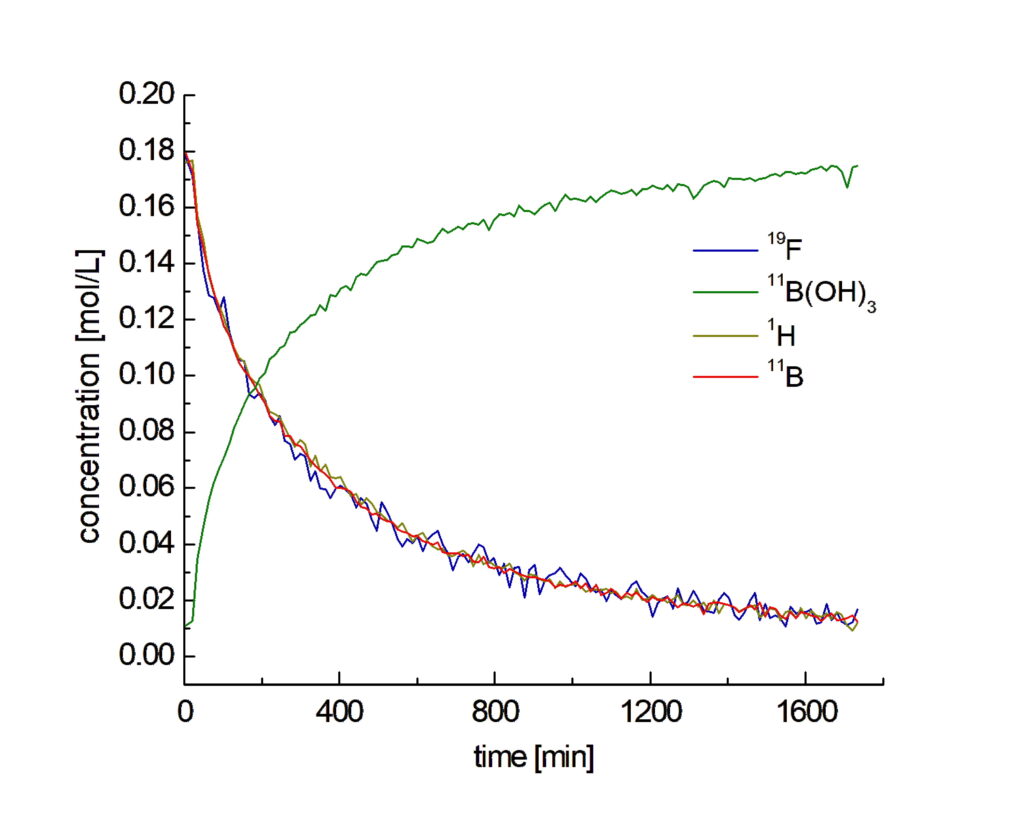
Figure 7: Concentrations of the different nuclei during the Suzuki-Miyaura-Coupling of 6-Fluoro-3-pyridinylboronic acid and 1-Bromo-4-fluorobenzene in aqueous phase.
Conclusion
In summary, in this application note we demonstrate that combining two Spinsolve spectrometers with two suitable in-line filters, it is possible to simultaneously monitor the species dissolved in the two immiscible phases of a biphasic reaction mixture. This is the very first time that starting materials, intermediates, byproducts, and product reacting at the interphase of two immiscible solvents could be monitored in real time by NMR to provide full understanding of the reaction kinetics. The concentration of the different species dissolved in both protonated solvents could be accurately monitored thanks to the good performance of the solvent suppression sequence available in the Spinsolve software.
The stability of the system and the ease of use of the software make it possible for non‑expert NMR users to carry out on‑line reaction‑monitoring experiments with minimal training. The use of PTFE tubing and connectors—commonly employed in HPLC systems—together with our flow cell installed inside the Spinsolve enables the reaction to be performed under inert conditions.
To get the complete App Note please Click Below
References
[1] A. Slattery, Z. Wen, P. Tenblad, J. Sanjose‑Orduna, D. Pintossi, T. den Hartog, T. Noël, Science, 2024, DOI: 10.1126/science.adj1817
[2] P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams, C. O. Kappe, Angew. Chem. Int. Ed. 2021, 60, 8139.
[3] M. Bornemann-Pfeiffer, J. Wolf, K. Meyer, S. Kern, D. Angelone, A. Leonov, L. Cronin, F. Emmerling, Angew. Chem. Int. Ed. 2021, 60, 23202.
[4] L. Schmidt, L. Jolly, L. Hennecke, F. Lopez Haro, H. Gröger, Andreas Liese, Organic Process Research & Development, 2024, 28, 3791, DOI: 10.1021/acs.oprd.4c00076
[5] a) C. Röhlich, A. S. Wirth, K. Köhler, Chemistry–A European Journal, 2012, 18, 15485-15494. b) M. Javaherian, P. Movaheditabar, Journal of the Iranian Chemical Society, 2023, 20, 2103-2125.
[6] Y. Sato, J. Liu, I. E. Ndukwe, M. V. S. Elipe, D. J. Griffin, J. I. Murray and J. E. Hein Chem Catal., 2023, 3, 100687.