In this short case study, we follow the outcome of a typical enantioselective CBS (Corey, Bakshi, Shibata) reduction reaction of acetophenone 3 to its corresponding alcohol 5 using both 11B and 13C NMR measurements on a Spinsolve Multi X NMR system. The stereoselective CBS reduction was first reported by E.J. Corey et al. in 1987.[1] They employed the chiral boron containing catalyst 2, which is the active catalytic species in the process. This moiety 2 first needs to be activated by adding it to a borane solution in THF (1M BH3*THF) starting from the commercially available pre-catalyst 1.

Scheme 1: Activation of the pre-catalyst 1 with 1M BH3*THF (5 min at 23°C) for the CBS reduction.
Afterwards, this activated species 2 serves as the catalyst in the reduction of acetophenone 3. As shown in scheme 2, the intermediate product of the reduction is compound 4, which can nicely be observed by 11B NMR. The final asymmetric alcohol product 5 can be obtained after an acidic work-up employing HCl in MeOH. For this study, we stopped our investigation after obtaining 4, which was made visible by 11B and 13C NMR.
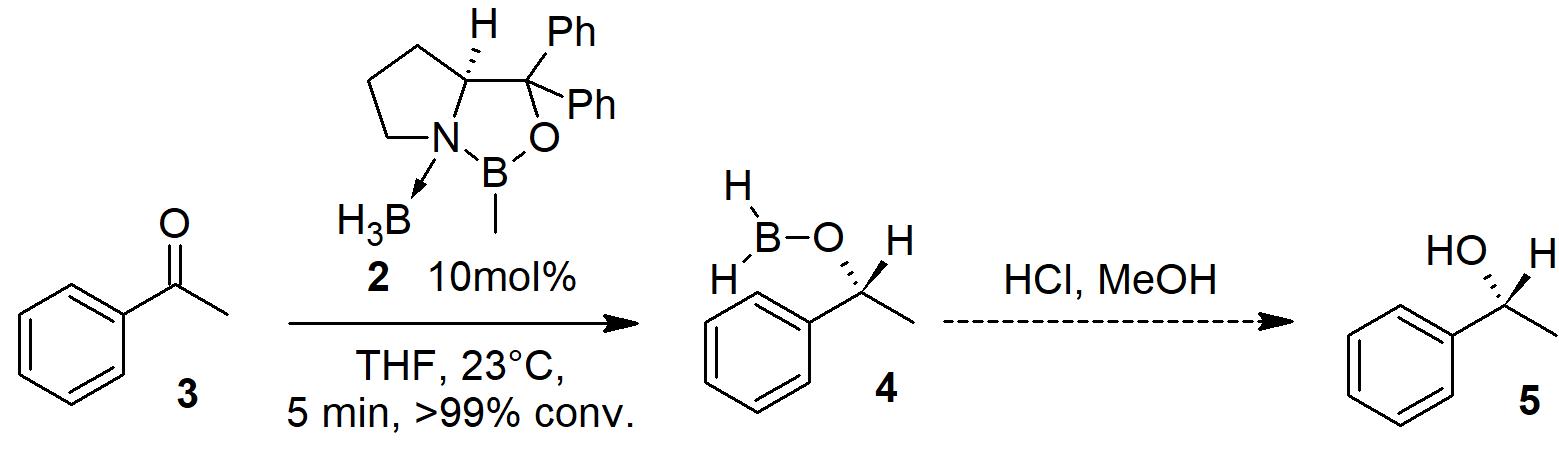
Scheme 2: CBS reduction of acetophenone 3 (50 mg; 0.4 mmol) employing the previously activated catalyst 2 (10 mol%) in 0.6 mL of 1M BH3*THF solution to the corresponding intermediate 4.
To follow the boron species involved in the catalytic CBS reduction, a Spinsolve Multi X was used with its X channel set to 11B. Figure 1 shows a stack plot of the different species:
a) CBS-Me pre-catalyst 1 shows a broad signal at around 8 ppm (orange)
b) The spectrum of the 1M BH3*THF solution shows the borane signal at around -1 ppm (blue)
c) Mixture of the free BH3*THF (blue), the –N-BH3 signal of the activated CBS-catalyst 2 (yellow) with a corresponding signal at around -15 ppm and a broad signal range at around 20-40 ppm (red) containing different signals of monomeric/dimeric B-Me signals of 1 and 2
All boron spectra were recorded with 1H decoupling on a 60 MHz Multi X Spinsolve spectrometer. The assignment of the 11B NMR signals is in agreement with the literature.[1]
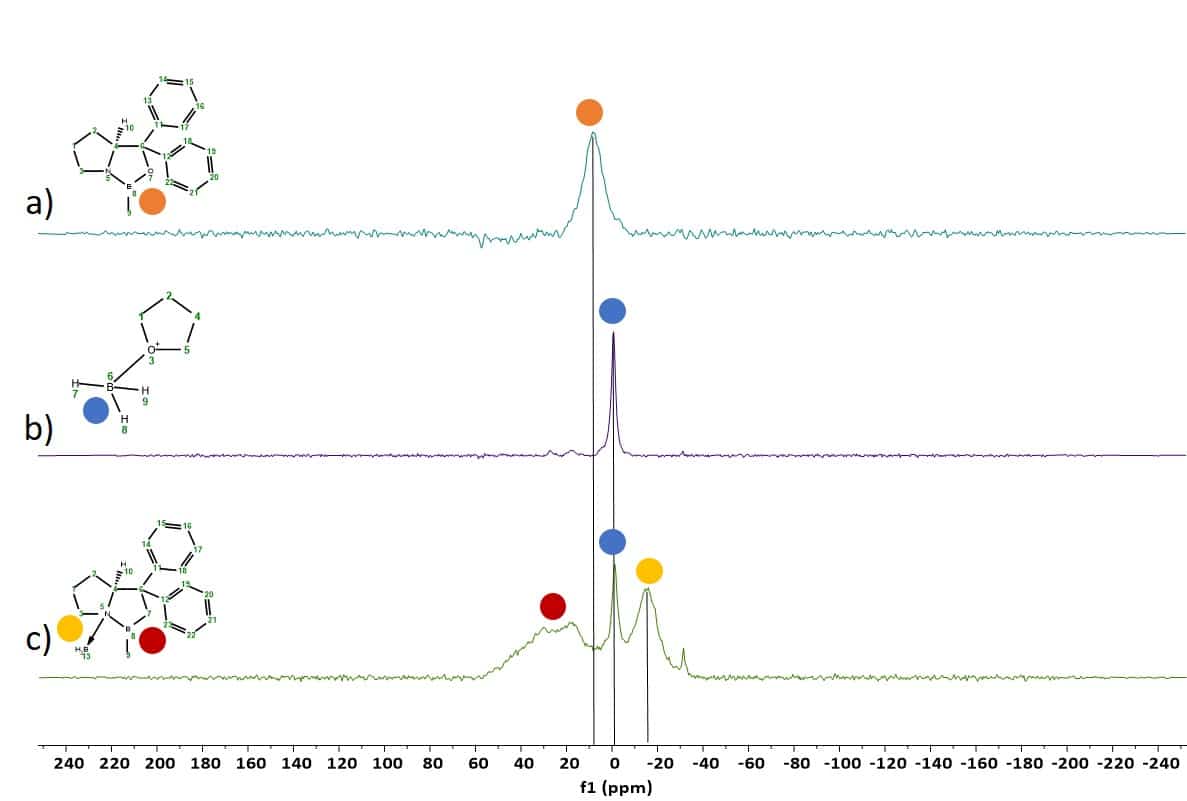
Figure 1: 11B spectra of the different boron species in the CBS reduction of acetophenone 3.
To check the result of the reaction, 13C NMR spectra before and after the reduction of acetophenone were acquired on the same Spinsolve Multi X by switching its X channel to 13C. The stacked spectra show the zoom of the aromatic region of the 13C spectra of a) the starting material acetophenone 3 and b) the boron intermediate 4 after the reaction. A clear difference between the spectra can be observed. The carbonyl peak 7 of acetophenone 3 is absent in the spectrum of the intermediate product 4. In addition, the signal for carbon 6 shifts towards a lower chemical shift value of 127 ppm. Moreover, the four aromatic CH signals 2-5 from the starting material 3 split into the signals 4-5 and 2-3 due to the presence of the boron moiety, which breaks the symmetry of the molecule up. Finally, the quaternary carbon 1 which is visible at 135 ppm in a) shifts to 145 ppm in the intermediate product spectrum b).

Figure 2: 13C NMR spectra of a) acetophenone 3 and b) CBS reduction intermediate product 4 all in THF measured on a Spinsolve 60 MHz Multi X spectrometer.
The completion of the reaction can also be verified by 11B NMR. Figure 3 shows the corresponding 11B spectrum after the reaction of acetophenone 3 with the activated CBS catalyst 2 showing the corresponding 11B peak of the –O-BH2 species at around 27 ppm (green). As a large excess of BH3*THF was employed, the respective signal (blue) is also visible after the reaction. The boron spectrum was recorded with 1H decoupling on a 60 MHz Multi X Spinsolve spectrometer. The 11B NMR data are in accordance with the literature.[1]

Figure 3: 11B NMR spectrum of the reaction mixture after the reaction of acetophenone 3 in THF measured on a Spinsolve 60 MHz Multi X spectrometer.
References:
[1] Corey, E. J.; Bakshi, R. K.; Shibata, S. J . Am. Chem. Soc. 1987, 109, 5551-5553.